Las enfermedades neurodegenerativas en la infancia son un grupo raro poco frecuente de enfermedades neurológicas que causan síntomas que resultan debilitantes de forma progresiva, además del deterioro irreversible del cerebro.1-3
Se presentan como una degeneración inesperada de la función cerebral. Este deterioro se asocia con mayor frecuencia a las enfermedades que padecen adultos, tal como el Alzheimer, y es común que sea desconocido y diagnosticado erróneamente cuando afecta a niños.1
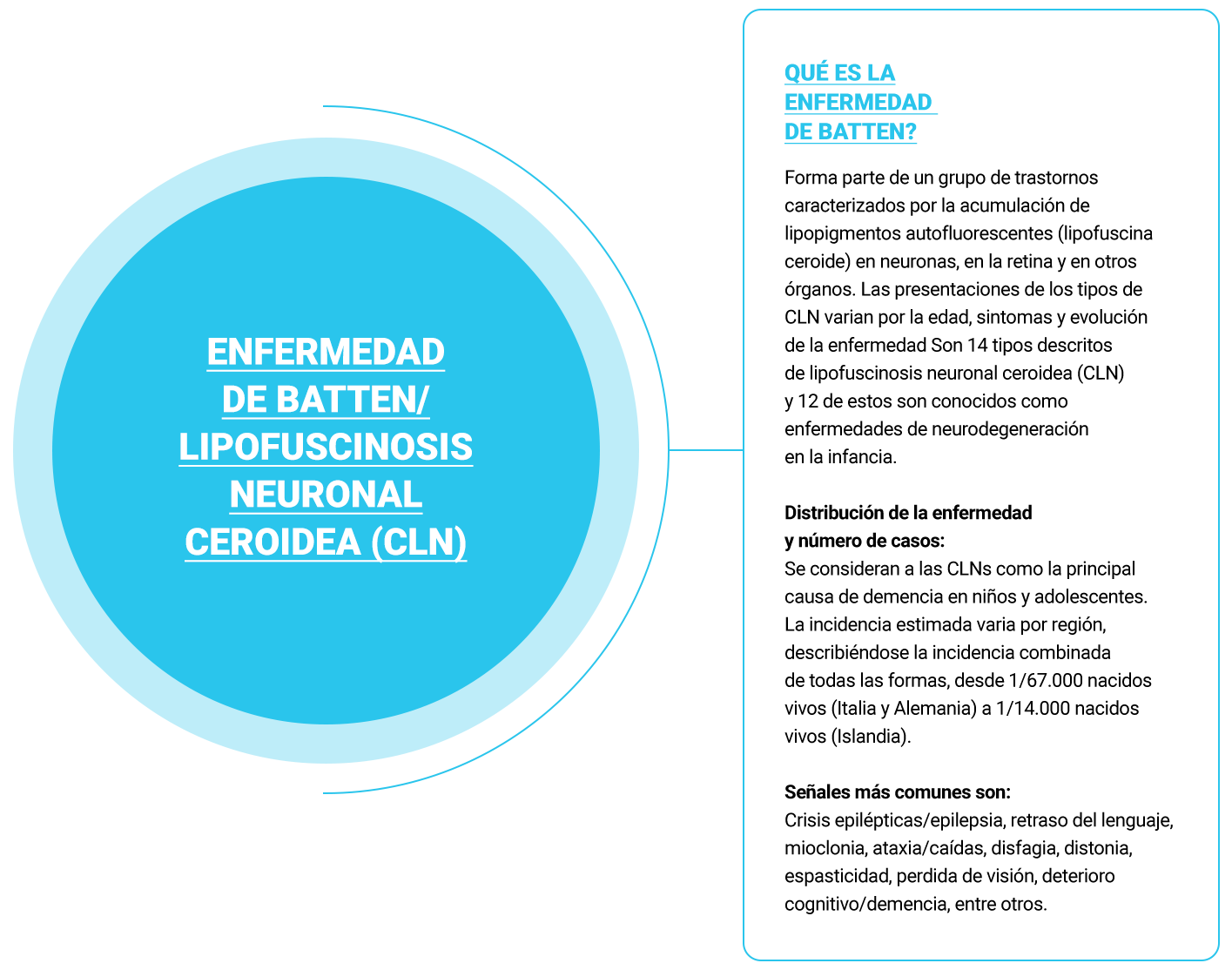
La enfermedad de Batten/Lipofuscinosis Neuronal Ceroidea hace parte del grupo de enfermedades neurodegenerativas en la infancia de origen genética.
Muchas formas de enfermedades neurodegenerativas en la infancia, que inicialmente se presentan como convulsiones recurrentes, progresan a daño cerebral irreparable, demencia, ceguera y pérdida de funciones motrices, tales como la capacidad de deglutir.1-3